Миоклонические припадки
Ювенильная миоклоническая эпилепсия
1. Как протекает болезнь? 2. Диагностика заболевания 3. Лечение
Ювенильная миоклоническая эпилепсия – форма эпилепсии, которая встречается преимущественно у детей. Ее дебют начинается в возрасте 13–18 лет, хотя может произойти гораздо раньше или позже (с 7 до 21 года). Основные симптомы болезни включают в себя миоклонические приступы, которые затрагивают парные органы, в основном – верхней части тела (глаза, плечи). В некоторых случаях судороги появляются на ногах и всех остальных частях тела.

Впервые признаки заболевания были описаны в 1867 году, но выделена в отдельный тип эта эпилепсия была в 1955 году Янцем с коллегами. Именно поэтому болезнь носит второе название – синдром Янца.
Симптомы болезни могут быть схожими с теми, которые наблюдаются у заболевания под названием «миоклонус-эпилепсия». Но это два отличающихся друг от друга заболевания, которые характеризуются разным течением и прогнозом.
Как протекает болезнь?
Обычно судороги у подростков возникают сразу после пробуждения, быстро проходят, не сопровождаются потерей сознания. Эти симптомы не рассматриваются больными, как серьезные, поэтому долгое время помощь не бывает оказана. Однако ювенильная миоклоническая эпилепсия без фармакологической поддержки практически не может войти в стадию ремиссии. Медицинское лечение назначается уже после того, как признаки дополняются генерализованным приступом.
Многие люди знают, что такое миоклонические судороги (подергивания крупных мышц тела). Они могут испытать их при засыпании или проснуться от их проявления. Такие единичные симптомы не являются проявлением какого-либо заболевания, являются физиологической нормой.
Часто к судорогам у детей впоследствии могут добавляться генерализованные припадки, процесс которых идентичен с теми, что возникают при «большой» эпилепсии, и могут носить развернутый характер или принимать малую форму (потеря сознания с судорогами без падений).

Ювенильная миоклоническая эпилепсия может сопровождаться, помимо судорог, абсансами. Такие приступы проявляются кратковременным застыванием человека без судорожных проявлений с открытыми глазами – в это время у него теряется сознание. Такие припадки у детей могут стать предвестниками основного заболевания – юношеской миоклонической эпилепсии.
Характерные симптомы, которые часто можно наблюдать при этой болезни у подростков – эмоциональная лабильность, плаксивость, сильная впечатлительность. При этом уровень интеллекта у них не страдает. Прогноз благоприятный при регулярно проводимой лекарственной терапии.
Диагностика заболевания
Врачу необходимо дифференцировать миоклоническую эпилепсию от других болезней и синдромов.
- Тикозные расстройства – являются самыми распространенными неврологическими нарушениями среди детей. Характеризуются стереотипными движениями, подергиваниями. Дисфункция нервной системы называется в основе причины тиков. Врач с помощью наблюдения и функциональных проб устанавливает правильный диагноз.
- Миоклонус-эпилепсия. Несмотря на схожее название и некоторые идентичные симптомы, эти заболевания имеют различные причины и течение. В строгом смысле слова, миоклонус-эпилепсия – это синдром, который проявляется при многочисленных серьезных заболеваниях, пограничных и острых состояниях. Основное отличие от миоклонической эпилепсии заключается в том, что неврологические расстройства прогрессируют в тяжелые состояния: деменцию, атаксию. Многие болезни, которые сопровождает миоклонус-эпилепсия, заканчиваются летальным исходом.
- Фокальные приступы при «большом» варианте эпилепсии. Они достаточно разнообразны и сложны по симптоматике. Некоторые из них человеку неосведомленному действительно могут напомнить миоклоническую эпилепсию.
Лечение
Медикаментозную терапию рекомендуют проводить постоянно. У более 90% пациентов, которые прекратили прием лекарственных препаратов, начиналось обострение. Самыми действенными веществами в терапии этого вида эпилепсии считаются вальпроаты. Однако из-за их возможной тератогенности представителям женского пола рекомендуют применять ламотриджин и леветирацетам. Может быть назначена комбинированная терапия.

При правильно подобранной терапии больные могут вести полноценную жизнь, эта форма эпилепсии с блокированными приступами не накладывает какой-либо отпечаток на общение, учебу, работу, создание семьи.
В настоящее время ученые ищут способы, которые позволят им эффективно бороться с эпилепсией, вызванной генной мутацией, непосредственно влияя на ее причины. Один из таких методов – трансфекция, которая предполагает ввод новых генов в клетки. Синтезируя белок, такие гены начинают оказывать лечебный эффект.
Итак, юношеская миоклоническая эпилепсия – это неврологическое нарушение, одна из форм эпилепсии, которая возникает у детей старшего возраста и у подростков. Она характеризуется миоклоническими судорогами верхней части тела, преимущественно сразу после пробуждения. Впоследствии к ним может присоединиться генерализованный припадок. В качестве основной причины называется наследственная предрасположенность. Прогноз заболевания благоприятный при адекватной терапии.
Здоровье, медицина, здоровый образ жизни
Миоклонические припадки
Этиология и патофизиология
Миоклонические судороги характеризуются повторными подергиваниями крупных —упп мышц и могут быть обусловлены рядом причин.
– Часто наблюдаются у пациентов с диффузным поражением головного мозга, затра–ивающим серое вещество: болезни накопления, инфекционные заболевания (например, болезнь Крейтцфельдта-Якоба, подострый склерозирующий панэнцефа-лит).
– Могут носить приобретенный характер, когда возникают на фоне метаболических нарушений (уремия, гипоксия, гиперосмолярные состояния, паранеопластические синдромы).
• Могут возникать на фоне прогрессирующих неврологических заболеваний: прог-зессирующая миоклоническая эпилепсия с тельцами Лафоры или без них.
• Могут возникать как проявление первичной генерализованной эпилепсии: юве–ильная миоклоническая эпилепсия или абсансы с миоклоническим компонентом.
• Часто встречаются у новорожденных и связаны у них с нейродегенеративными состояниями (например, ганглиозидозами: болезнь Тея-Сакса, болезнь Альперса).
• Приобретенные миоклонические судороги возникают в любом возрасте.
Дифференциальный диагноз
• Тремор.
• Миокимия.
• Очаговые моторные судороги.
• Тик.
• Тетания.
• Доброкачественные миоклонические подергивания при засыпании.
• Гиперреакция при испуге.
Симптоматика
• Миоклонические подергивания – короткие клонические движения, возникающие спорадически в различных частях тела, могут быть ограничены одной конечностью, могут иметь более распространенный характер, включая и осевую мускулатуру.
• Мультифокальный миоклонус возникает чаще всего при метаболических заболеваниях, гипоксии или уремии.
• Миоклонические судороги могут постепенно переходить в тонико-клонические, так происходит, например, при ювенильной миоклонической эпилепсии.
Диагностика
• Наблюдение кратковременных миоклонических подергиваний или описание таковых самим больным либо свидетелем.
Электроэнцефалография помогает диагностировать первичную генерализованную эпилепсию с миоклоническим компонентом.
– Электроэнцефалография также позволяет исключить диагноз эпилептического миоклонуса, если на фоне миоклонических подергиваний не наблюдается никаких ЭЭГ-изменений.
• Для исключения метаболических причин миоклонуса проводится биохимическое исследование крови.
• Визуализирующие исследования показаны, когда другими методами не удается установить причину заболевания.
Лечение
• Терапевтический эффект оказывают многие препараты, для кратковременного контроля проявлений заболевания лучше всего подходят бензодиазепины.
• При первичной генерализованной эпилепсии с миоклонусом наиболее эффективным препаратом является вальпроат, другие возможные варианты: топирамат, ламотриджин, зонисамид и леветирацетам.
• Для полного устранения миоклонуса часто необходима коррекция основного заболевания, в том числе метаболических нарушений.
Важные замечания
• Прогноз зависит от этиологии миоклонуса.
– Прогноз крайне неблагоприятный, если миоклонус является симптомом генерализованного нейродегенеративного заболевания.
– Если миоклонус связан с метаболическими нарушениями (например, уремией), коррекция основного заболевания часто приводит к существенному облегчению неврологической симптоматики.
• Первичная генерализованная эпилепсия с миоклонусом часто с успехом лечится вальпроатом.
Миоклонические припадки
При некоторых эпилепсиях старшего детского и подросткового возраста миоклонии составляют преобладающий, если не исключительный тип припадков. Эпилепсии, характеризующиеся выраженными миоклоническими феноменами, в этом возрастном промежутке в большинстве случаев идиопатические по происхождению. Очень часто наблюдается выраженная генетическая предрасположенность к судорожным расстройствам (Arzimanoglou et al., 2004).
Более типичный в старшем детском и подростковом возрасте феномен — последовательность миоклонических судорог, часто перерастающая в генерализованный тонико-клонический припадок (клонически-тонико-клонический припадок). Течение и лечение миоклонических атак, описываемых в этом разделе, отличаются от течения и лечения синдромов Леннокса-Гасто и Драве, описанных в предыдущих разделах. Важно правильно распознать тип припадка, для этого требуется не только подробный опрос родителей, но также при наличии показаний выполнить полиграфию и/или видео-ЭЭГ.
Также важно точно определить другие типы припадков, обычно сопровождающих миоклонические атаки, так как именно сопутствующие припадки являются ключевым признаком классификации миоклонических эпилепсий.
Эпилептические синдромы с выраженным миоклонусом можно разделить на три основные категории:
1. те, при которых миоклонусу сопутствуют в основном абсансы (эпилепсия с миоклоническими абсансами и миоклония век с абсансами);
2. те, при которых миоклонические атаки избирательно индуцируются прерывистой фотостимуляцией;
3. эпилепсии, начинающиеся в подростковом возрасте, при которых основным типом припадков является grand mal, но могут также наблюдаться и абсансные атаки (т.е. ювенильная миоклоническая эпилепсия, обсуждаемая ниже);
4. эпилепсии, дебютирующие в широком возрастном диапазоне, от старшего детского до взрослого возраста, с наибольшей выраженностью наследственного фактора, с выраженным ритмичным дистальным миоклонусом и генерализованными судорогами, и тонико-клоническими и фокальными припадками у некоторых пациентов. Duron et al. (2005) проанализировали частоту различных типов припадков при генерализованном эпилептическом синдроме, их зависимость от возраста, и временной профиль припадков.
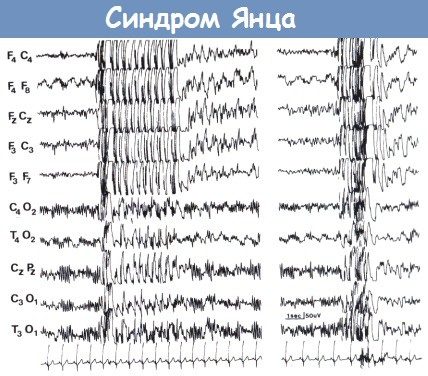 Ювенильная миоклоническая эпилепсия (синдром Янца).
Ювенильная миоклоническая эпилепсия (синдром Янца).
Слева видны быстрые (4 Гц) разряды спайк-волн с полиспайками в начале (без клинической корреляции).
Справа показаны миоклоническая судорога (виден миографический артефакт на ЭКГ) и разряды полиспай-ков на ЭЭГ.
а) Ювенильная миоклоническая эпилепсия (ЮМЭ). ЮМЭ (также называемая миоклонической эпилепсией подросткового возраста или синдромом Янца) — наиболее частый и узнаваемый синдром идиопатической генерализованной эпилепсии. Заболевание начинается между 12 и 18 годами жизни (Delgado-Escueta и Enrile-Bacsal, 1984; Janz, 1989), но может дебютировать и вне этогих возрастных рамок. Иногда это состояние называют «миоклонический petit mal» или «импульсивный petit mal», что вносит путаницу, так как оно совершенно отлично от абсанс эпилепсии. ЮМЭ встречается у 5-11% подростков с эпилепсией. Миоклонические судороги поражают в основном плечи и руки, редко — нижние конечности.
Они могут быть асимметричными и даже односторонними (Genton et al., 1994; Janz, 1994). Сознание обычно сохранено, но из-за непроизвольных движений пациент часто роняет предметы, которые держит в руках во время припадка.
Судороги возникают в основном после пробуждения и могут быть единичными сокращениями или серийными судорогами, однако редко достигающими состояния миоклонического статуса. У 90% пациентов наблюдаются сопутствующие генерализованные тонико-клонические припадки. Иногда они могут развиваться вслед за сериями судорог, такую последовательность называют клонико-тонико-клоническим припадком. От 15 до 30% пациентов также имеют абсансы. Сложные взаимосвязи между различными идиопатическими эпилепсиями обсуждались Thomas et al. (2005).
Типичная иктальная ЭЭГ состоит из вспышек спайков высокой частоты, за которым следует одна или несколько медленных волн. Такие же комплексы полиспайк-волна могут наблюдаться в интериктальный период. Фокальные ЭЭГ-проявления могут присутствовать почти в 20% случаев (Panayiotopoulos, 1994а). Лишение сна и, часто, фотостимуляция, могут провоцировать припадки.
Около 80% пациентов хорошо реагирует на терапию вальпроатом натрия, но курс лечения необходимо продолжать, возможно, неопределенно долго. У женщин с ЮМЭ при выборе наиболее подходящего АЭП нужно учитывать возможность предстоящей беременности (Tomson и Battino, 2005). В резистентных случаях нужно попытаться назначить бензодиазепины. Также применялся ламотриджин, с вариабельным эффектом, включая ухудшение или отсутствие контроля миоклонических судорог в некоторых случаях. Топирамат и зонисамид являются интересными альтернативными препаратами. Рандомизированное двойное слепое контролируемое исследование выявило явную эффективность леветирацетама для контроля миоклонических судорог при ЮМЭ (Verdu et al., 2005).
Специфический профиль фармакочувствительности при ЮМЭ, совпадающий с профилем других идиопатических генерализованных эпилепсий, указывали Thomas et al. (2006); они подчеркивали потенциальную возможность усиления припадков при использовании фенитоина и особенно карбамазепина, который даже может спровоцировать миоклонический статус. Очень важным фактором в предотвращении припадков, вызываемых лишением сна, является устранение причин, препятствующих физиологическому сну.
Реакция на лечение отличная или хорошая, полный контроль припадков достигается в 80-90% случаев; спонтанная ремиссия, как сообщается, наступает очень редко. С другой стороны, рецидив припадков после прекращения лечения случается часто, даже после контроля припадков в течение многих лет, но в некоторых случаях может задержаться на несколько лет (Thomas et al., 2005).
Диагноз обычно не вызывает трудностей. Однако необходимо исключать редкие прогрессирующие миоклонические эпилепсии, такие как болезнь Лафора и некоторые случаи болезни Унферрихта-Лундборга, так как в начале заболевания они могут имитировать ЮМЭ. Последовательное ухудшение миоклонического синдрома, появление медленной фоновой ЭЭГ-активности и деградация когнитивных функций предполагает вероятность наличия прогрессирующего заболевания (Arzimanoglou et al., 2004).
Генетические факторы играют значительную роль в этиологии этого синдрома. Было выполнено несколько исследований (подробнее см. Thomas et al., 2005), предполагается связь с коротким плечом 6 хромосомы или длинным плечом 15 хромосомы. Результаты семейных исследований, в основном касающиеся механизма наследования, часто противоречивы. Существующая полигенная модель считается наиболее вероятной (Bate и Gardinger, 1999), и может объяснить неоднозначные и противоречивые результаты исследований связи с хромосомами, основанных на упрощенных менделевских моделях наследования.
Такая полигенная модель предполагает наличие общей центральной группы генов, вызывающих снижение эпилептогенного порога при идиопатических генерализованных эпилепсиях, в этом случае специфические проявления ЮМЭ явились бы следствием действия одного или нескольких других генов (Sander et al., 2000).
б) Миоклония век с абсансами. Это расстройство характеризуется очень частым возникновением судорог век с отведением глазных яблок вверх, которым может сопутствовать короткий период легкого отсутствия восприятия (Jeavons, 1982; Appleton et al., 1993; Panayiotopoulos, 1998). На ЭЭГ выявляются разряды коротких (менее 6 секунд) комплексов полиспайк-волна, отмечается выраженная фоточувствительность. Эффективно лечение вальпроатом натрия и другими АЭП, показанными при идиопатических эпилепсиях, но, возможно, лечение должно продолжаться и во взрослом возрасте.
в) Другие миоклонические синдромы. Они включают поздние случаи, идентичные «доброкачественной миоклонической» эпилепсии, с поздним началом в возрасте до 5-6 лет (Guerrini et al., 1994а) и самопроизвольные фотомиоклонические припадки.
Наследуемые по аутосомно-доминантному механизму кортикальный тремор, миоклонус и эпилептические припадки, наблюдавшиеся во многих японских и европейских семьях, описывались под разными названиями, сейчас они известны как семейная миоклони-ческая эпилепсия взрослых. Striano et al. (2005) изучили семейные случаи с такой клинической картиной и заключили, что, несмотря на их генетическую гетерогенность, все они представляют одно клиническое состояние, в японских семьях сцепленное с 8q24, а в итальянских — с 2р11.1-q12. Может быть задействован и третий локус, были зафиксированы спорадические случаи с такими же характеристиками.
Редактор: Искандер Милевски. Дата публикации: 4.1.2019
МИОКЛОНУС-ЭПИЛЕПСИЯ
Миоклонус-эпилепсия (греч. mys, my [os] мышца + клонус; эпилепсия; син.: прогрессирующая миоклоническая эпилепсия, болезнь Унферрихта — Лундборга) — наследственное семейное заболевание центральной нервной системы, основным клиническим проявлением к-рого является сочетание миоклонического гиперкинеза с эпилептическими припадками. Описана Унферрихтом (H. Unverricht) в 1891 г. и Лундборгом (H. В. Lundborg) в 1903 г. как отдельная нозологическая форма.
Содержание
Этиология и патогенез
Заболевание наследуется по аутосомно-рецессивному типу. В основе его, по-видимому, лежит врожденное нарушение обмена веществ, но первичный биохимический дефект еще не выявлен. Оба пола заболевают одинаково часто. С. Н. Давиденков (1936) считал, что сложная картина Миоклонус-эпилепсии является выражением своеобразного плейотропизма гена этой болезни. Родители больных Миоклонус-эпилепсией могут быть здоровыми (нередки случаи их кровного родства). У родственников больных М.-э. наблюдаются и другие заболевания ц. н. с. — эпилепсия, паркинсонизм, хорея, умственная отсталость, алкоголизм и др. Описаны изоляты с относительно высокой частотой М.-э. Наряду с семейными наблюдается немало спорадических случаев.
Патологическая анатомия
При патологоанатомическом исследовании в головном мозге обнаруживаются дистрофические изменения, наиболее выраженные в зубчатом ядре мозжечка, оливах продолговатого мозга, черном веществе, полосатом теле, таламусе (зрительном бугре), в разных участках коры мозга. Характерным, но не обязательным гистопатологическим признаком М.-э. является наличие одного-двух шарообразных амилоидных включений преимущественно в нервных клетках, к-рые были открыты Лафорой (G. R. Lafora, 1911) и названы его именем. Тельца Лафоры обнаруживаются не только в ганглиозных клетках головного и спинного мозга, но также и в корешках спинномозговых нервов, в периферических нервах, скелетных мышцах, печени, селезенке. Во многих нервных клетках имеются также значительные скопления липофусцина.
Клиническая картина
В большинстве случаев болезнь начинается в возрасте 10—15 лет. Для нее характерно сочетание миоклонического гиперкинеза с эпилептическими припадками. У одних больных первыми симптомами являются миоклонии (см.), а общие эпилептические припадки (см. Эпилепсия), обычно ночные, присоединяются позже, нередко спустя ряд лет. У других больных вначале наблюдаются только эпилептические припадки, без миоклонического гиперкинеза, к-рый появляется позже. Кроме больших эпилептических припадков с потерей сознания, клонико-тоническими судорогами, недержанием мочи, прикусом языка, могут возникать и другие проявления эпилепсии — малые припадки, абсансы (разновидность малого эпилептического припадка), сумеречные состояния и др.
При М.-э. миоклонический гиперкинез характеризуется большим разнообразием и имеет свои особенности. Первоначально возникающие клонические подергивания являются чаще изолированными, могут наблюдаться в одной мышце; преимущественно сокращается четырехглавая мышца бедра, с незначительным двигательным эффектом. В дальнейшем миоклонии становятся все более распространенными, захватывают мышцы конечностей, туловища, головы, увеличивается их частота и двигательный эффект.
В большинстве случаев наблюдаются миоклонии быстрого темпа, молниеносные, беспорядочные, неритмичные, асинхронные, нерегулярные. Они значительно уменьшаются и прекращаются в спокойном состоянии, в лежачем положении, отсутствуют во время сна. Их усиливают произвольные движения и эмоции, особенно отрицательные. Наблюдаются так наз. сенсоклонические реакции — неожиданно возникший яркий свет, громкий звук вызывают кратковременный миоклонический приступ в виде внезапного усиления мышечных сокращений в конечностях, туловище, лице с резкими общими вздрагиваниями.
Миоклонический гиперкинез проявляется в различной степени в разные дни — он может значительно затихать или, наоборот, усиливаться, в связи с чем сами больные отмечают у себя чередование «хороших» и «плохих» дней. Иногда у нек-рых больных появляются психотонические приступы, описанные Лундборгом. Они выражаются в том, что под влиянием эмоциональных воздействий у больного возникает кратковременное тоническое напряжение всей мускулатуры без утраты сознания.
Описаны редко встречающиеся сочетания миоклонического гиперкинеза с хореоатетозом (см. Гиперкинезы). Имеется тесная связь между миоклоническими гиперкинезами и эпилептическими припадками. Изо дня в день усиливающиеся миоклонии постепенно становятся особенно резко выраженными, на высоте которых больной на короткое время теряет сознание, появляются клонико-тонические судороги с прикусом языка и недержанием мочи. После окончания эпилептического припадка сразу исчезают миоклонии, обычно на один-два дня, а затем они появляются вновь и постепенно усиливаются. Миоклонус-эпилепсия имеет прогрессирующее течение. Из-за миоклоний у больных затруднена и постепенно становится невозможной ходьба и выполнение других произвольных движений, они не могут себя обслуживать, самостоятельно принимать пищу. У отдельных больных наблюдаются легкие мозжечковые расстройства (атаксия, интенционное дрожание), мышечная гипотония. Чувствительность сохранена. Парезов, рефлекторных нарушений не отмечается. В поздних стадиях болезни развивается Экстрапирамидная обездвиженность с клин, картиной паркинсонизма (см.), при этом исчезает миоклонический гиперкинез.
За небольшими исключениями, больные деградируют психически.
На ранних этапах заболевания отмечаются астеноневротические расстройства, в дальнейшем больные становятся все более вялыми, апатичными, снижается интерес к окружающему, ухудшается память, ослабевает сообразительность, критика, временами вялость сменяется раздражительностью, бывают состояния спутанности с галлюцинаторными переживаниями, у многих развивается тяжелая деменция (см. Слабоумие). При электроэнцефалографическом исследовании (см. Электроэнцефалография) часто обнаруживаются пароксизмы электрической активности высокой амплитуды. Световая стимуляция, гипервентиляция усиливают пароксизмальную электрическую активность мозга. В сыворотке крови больных часто обнаруживается уменьшение содержания мукополисахаридов. По данным нек-рых авторов, для М.-э. характерна также повышенная концентрация в крови аргининсукцининовой к-ты.
Диагноз
Диагноз ставится на основании сочетания в клин, картине миоклонического гиперкинеза с эпилептическими припадками. Для диагностики важное значение имеет семейный характер заболевания. Дифференциальный диагноз проводится с весьма сходной мозжечковой миоклонической диссинергией Ханта — наследственным, аутосомно-рецессивным заболеванием, при к-ром также наблюдаются миоклонический гиперкинез и эпилептические припадки, но, в отличие от М.-э., мозжечковые расстройства гораздо более выражены. В отличие от М.-э., при кожевниковской эпилепсии (см.) клонические судороги наблюдаются в определенной части тела и обычно не носят генерализованного характера. Особенно трудно установить ранний диагноз в спорадических случаях, когда заболевание проявляется неполным клин, синдромом — или эпилептическими припадками, или миоклоническим гиперкинезом. В этом случае имеет значение семейный анамнез, а также уменьшение содержания мукополисахаридов в сыворотке крови.
Лечение и Прогноз
Лечение симптоматическое. Применяется противосудорожная терапия, назначают фенобарбитал, бензонал, седуксен, хлоралгидрат. Последние два средства могут значительно уменьшить на короткое время миоклонический гиперкинез. Применение хлоралгидрата может вызвать привыкание к нему и быстрое развитие кахексии, Показаны также повторные длительные курсы лечения глутаминовой к-той.
Прогноз неблагоприятный. Средняя продолжительность болезни ок. 20 лет, нек-рые больные доживают до старческого возраста. Наиболее часто причиной смерти является нарастающая кахексия, нередко пневмонии и другие интеркуррентные заболевания.
Супругам, в семье к-рых есть больной Миоклонус-эпилепсией, для решения вопроса о рождении ребенка рекомендуется обратиться в медико-генетическую консультацию (см.).
Библиография: Давиденков С. Н. Миоклонус-эпилепсия, в кн.: Неврол. и генетика, под ред. С. Н. Давиденкова, т. 2, с. 195, М., 1936; Дзержинский В. Myoclonia Unverricht’a, Журн, невропат, и психиат., кн. 5-6, с. 293, 1910; Маккьюсик В. А. Наследственные признаки человека, пер. с англ., М., 1976; Мельников С. А. Миоклонус-эпилепсия как синдром при некоторых заболеваниях головного мозга, Журн, невропат, и психиат., т. 57, в. 6, с. 740, 1957, библиогр.; de Ajuriaguerra J., Sigwald J. et Piot C. Myoclonie-epilepsie familiale de type Unverricht, Presse med., p. 1813, 1954; Bogaert L. Sur l’epilepsie-myoclonie progressive d’Unverricht — Lundborg, Mschr. Psychiat. Neurol., Bd 118, S. 170, 1949, Bibliogr.; Davison Ch. a. Keshner M. Myoclonus epilepsy, Arch. Neurol. Psychiat. (Chic.), v. 43, p. 524, 1940; Gambetti P. a. o. Myoclonic epilepsy with lafora bodies, Arch. Neurol. (Chic.), v. 25, p. 483, 1971; Hallidaу A. M. Les differents types des myoclonies, Rev. neurol., t. 119, p. 135, 1968; Handbook of clinical neurology, ed. by P. J. Yinken a. G. W. Bruyn, v. 15, p. 121, 1974, v. 27, p. 171, Amsterdam a. o., 1976; Lafora G. R. u. Glueck B. Beitrag zur Histopathologie der myoclonischen Epilepsie, Z. ges. Neurol. Psychiat., Bd 6, S. 1, 1911, Bibliogr.; LundborgH. Die progressive Myoclonus-Epilepsie, Up-sala, 1903; Unverricht H. Die Myoclonie, L]3z.— Wien, 1891.