Гипертрофия икроножных мышц
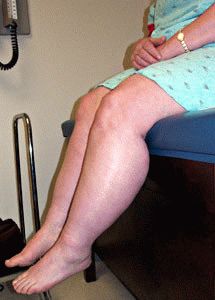
Атрофия мышц ног
Заболевание представляет редкую тяжёлую патологию, сопровождаемую мучительными проявлениями для больного. Из-за происходящих в организме человека патологических изменений происходит истончение и деформация скелетной мышцы. Мускулатура замещается тканью, не несущей способности сокращаться. Процесс называется атрофией мышц ног. Итогом болезни выступает утрата полноценной возможности двигаться, при полном перерождении тканей человек утрачивает её абсолютно. Важно начать полноценное лечение пациента вовремя.
Причины возникновения болезни
На начало развития патологии, связанной с атрофией мышц ног, воздействуют многочисленные аспекты, включающие:
- Понижение уровня метаболизма, сопровождается естественным старением организма человека;
- Всевозможные заболевания эндокринной системы, возникающий в итоге гормональный дисбаланс в работе организма;
- Трудности с пищеварительной системой;
- Наличие заболеваний соединительных тканей;
- Перенесение полиневритов;
- Последствия отдельных перенесённых инфекционных болезней и заболеваний, связанных с деятельностью паразитов;
Атрофия четырехглавой мышцы бедра чаще возникают после перенесённых операций и травм.
Симптомы заболевания
На первоначальном этапе человек замечает утомляемость и мышечную слабость в ногах, возникающую после физической нагрузки. Икроножные мышцы ног становятся заметными. Атрофии в первую очередь подвергаются проксимальные группы мышц ног. Процесс выражается в ограничении двигательной способности. К примеру, человеку становится трудно ходить по лестнице.
Мышцы ног атрофируются медленно, процесс растягивается на годы. Поражаются одна либо обе стороны, нося симметричный или асиметричный характер.
Любые симптомы болезни находится в тесной связи с возрастом пациента, общего состояния, характера развития формы заболевания. Сказанное отражается на выборе лечения.
- Постоянно нарастающая слабость мышц ног.
- Тремор.
- Неприятные ощущения (к примеру, чувство скребущих под кожей насекомых).
Наиболее ярким сигналом, указывающим на развитие атрофии, выступает существенное уменьшение мышцы, в которой развивается процесс. Причём уменьшение становится заметным даже на раннем этапе.
Заболевание считается хроническим. По ходу течения выделяют рецидивы, когда ощущается сильная болезненность в больной мышце. Случается ремиссия, однако симптомы лишь незначительно перестают беспокоить пациента.
Выделяют первичную атрофию мышцы, когда поражается собственно мускулатура и двигательные нейроны. Процесс связан с перенесёнными травмами либо отягощённой наследственностью.
Характеризуется быстрым наступлением утомления, потерей тонуса и начинающимися непроизвольными подёргиваниями ног.
При вторичной атрофии подвергаются патологическим процессам мышцы стоп и голени – участки деформируются, походка больного человека заметно меняется, сопровождаемая специфическим поднятием колен. Чаще процесс связан с перенесёнными заболеваниями инфекционного характера либо в связи с наследственной отягощённостью. Прогрессируя, заболевание способно перейти на верхнюю часть туловища. Наблюдается полный либо неполный паралич.
Указанная стадия атрофии делится на виды:
-
Прогрессирующая атрофия. Выявляется, благодаря симптомам, в детстве. Описанный вид заболевания имеет тяжёлое протекание, сопровождаясь резкими спадами артериального давления. Рефлексы сухожилий утрачиваются, при сохранении подергиваний конечностей, не связанных с желанием пациента;
Красноречивым признаком атрофии выступает уменьшение объёма больной мышцы, явление становится заметно по сравнению со здоровыми участками тела.
Миотония, протекающая на фоне атрофии мышц ног
В качестве заболевания выступает псевдогипертрофический вид Дюшенна. Заключается в частом проявлении миопатии, напрямую зависит от пола, проявляется исключительно у мальчиков.
Патология поражает организм детей в первые 5 лет жизни. Для заболевания характерна атрофия мышц таза и ног. Происходит раннее развитие псеводгипертрофий, включая мышцы икроножной зоны. Проверяя коленные рефлексы, можно отметить, что сухожилия подверглись ретракции. Ребёнок испытывает сложности в передвижении, не может прыгнуть либо нормально подняться по лестнице. Заболевание сопровождается развивающейся слабостью, мышцы плеч дополнительно включают в процесс атрофии. Спустя время ребёнок не сможет подниматься с постели.
Дальнейшие симптомы заболевания, если отсутствовало достаточное своевременное лечение, выражаются в проявлении заметной контрактуры из-за ретракции сухожилий. Развивается «конская» стопа.
Опасным проявлением заболевания выступает его влияние на мозг ребёнка, из-за чего мальчик начинает отставать в развитии. Изменяются мышцы сердца, ослабевает дыхательная система, сопровождаясь некачественной вентиляцией лёгких, часто развивается пневмония. Из-за патологического состояния органов, включая сердце и сердечную мышцу, пневмония проходит тяжело, периодически заканчиваясь летальным исходом.
В двадцатом веке учёный по имени Беккер смог описать доброкачественный вид миопатии, в дальнейшем обретший его имя.
Особенность заболевания заключается в проявлении после двадцати лет. Собственно атрофия протекает медленно, охватывая мышцы таза и бёдер. Характерной особенностью типа становится неизменность интеллектуальных способностей человека. Подобные типы патологии связаны с повреждением разных генов, находящихся в двух локусах Х-хромосомы, выступая в качестве генокопий.
Отметим – в одной семье не встречаются одновременно обе формы болезни.
Диагностика атрофии мышц ног
Диагностирование атрофии мышц ног проводится путём сбора детального анамнеза о человеке, о наличии хронических заболеваний и наследственной отягощённости. Пациента следует направить на сдачу развёрнутого анализа крови, чтобы определить уровень СОЭ, печёночных проб, глюкозы. Проводится процедура электромиографии.
Чтобы подобрать оптимальное лечение, медики назначают биопсию нервов и мышц. Попутно проводятся дополнительные исследования, если у пациента в анамнезе значатся хронические или обусловленные наследственностью заболевания.
Лечение заболевания
Лечение рассматриваемой патологии зависит от особенности протекания, форма, значение играет возраст больного.
Абсолютное излечение болезни невозможно, не существует специальных препаратов. Однако для нормализации жизни пациента применяют разнообразные методы медикаментозного лечения, направленные в первую очередь на снятие симптомов, чтобы улучшить обменные процессы в организме человека.
В конкретном случае доктор назначает индивидуальное лечение, опираясь на особенности больного. Единого, универсального подхода к лечению атрофии мышц ног не разработано.
В комплекс общего лечения входят витамины В и Е, препараты Дибазол, Прозерин и прочие. В редких случаях эффективной становится трансфузия крови.

Чрезвычайно важное значение при атрофии мышц ног играет массаж. Он поддерживает мышечный тонус, в значительной степени убрав симптомы заболевания, улучшает приток крови в сосуды пострадавшей конечности. Массаж позволяет быстро регенерировать мышечной ткани, обеспечивать клеточное дыхание тканей. Массаж выполняется часто, не реже раза ежедневно без перерывов. Каким образом делать массаж, сколько потратить времени, определяется соответствующим врачом в зависимости о тяжести состояния больного, особенностей мышц ног и протекающей атрофии.
Эффективным массаж становится при применении в случае послеоперационной атрофии. Процедура выполняется исключительно медицинским работником, знающим тонкости техники.
Необходимо делать неглубокий массаж, работа с мышцами должна быть аккуратной, без резких движений. При заболевании применяется общий массаж с простукиванием, непроникающий лёгкий массаж.
Нельзя забывать об использовании гимнастики, электрофореза, физиолечения. Гимнастика должна проводиться под наблюдением медика.
Терапевтическое лечение должно длиться постоянно, без перерывов, постоянно повторяться в соответствии с предписаниями докторов. Особо важным считается полноценное витаминизированное питание.
Псевдогипертрофическая миодистрофия Дюшенна (заболеваемость лиц женского пола)
В литературе имеются указания на заболевание миодистрофией Дюшенна лиц женского пола. При Х-сцепленном типе наследования теоретически это возможно при условии, если отец болен, а мать является гетерозиготным носителем. Однако практически для болезни Дюшенна такой вариант полностью отпадает, поскольку фертильность больных мальчиков равна нулю, большинство их не переживает пубертатный период.
Возможным вариантом заболевания девочки миодистрофией Дюшенна является хромосомная аберрация с моносомией по Х-хромосоме (Х0 — синдром Шерешевского — Тернера). Диагностика этого синдрома в раннем детском возрасте не всегда легка. Представляет интерес следующее наше наблюдение.
Больная С., 5 лет. Родилась от 5-й беременности, протекавшей с частой рвотой, тошнотой в течение всего срока, умеренным повышением уровня белка в моче, умеренным повышением АД. На 8-м месяце беременности у матери начались схватки, возникло кровотечение, она была госпитализирована. Роды на 10-й день после госпитализации. Шевеление плода в период беременности было нормальным. Девочка родилась с массой тела 2500 г, закричала сразу.
Головку начала держать с 3 — 4 мес, сидеть — с 7 мес, ходить — в 1 год 1 мес, при этом родители сразу же отметили особенность походки — девочка наступала в основном на пальцы стоп. В 4-летнем возрасте была заподозрена миодистрофия, и для уточнения диагноза она была госпитализирована в клинику. Четкого нарастания двигательного дефекта родители не отмечают, она несколько отстает от сверстников в беге, при вставании с пола прибегает к минимальным вспомогательным приемам.
Объективно: масса тела 20 кг 600 г. Умеренно выражен гипертелоризм, незначительная ретрогнатия. Шея несколько укорочена. Отсутствие талии. Умеренная брадикардия, нечистота I тона.
Интеллект грубо не снижен, однако контакт с девочкой затруднен, на большинство вопросов чаще всего реагирует инфантильной улыбкой, на вопросы отвечает не сразу.
Черепные нервы без особенностей. Сухожильные рефлексы не вызываются, брюшные рефлексы живые. Некоторая плотность икроножных мышц, умеренно выраженные ретракции ахилловых сухожилий. При исследовании мышечной силы отмечается минимальное снижение ее в проксимальной мускулатуре рук и ног. При ходьбе и беге тенденция наступать на пальцы. Фасцикуляции отсутствуют.
Миодистрофии Дюшенна у больной С-вой

При наличии мозаицизма ХО/ХХ (синдром Шерешевского — Тернера).
Видна короткая шея (а), псевдогипертрофия
икроножных мышц (б, в).
На рентгенограмме шейного отдела позвоночника отмечаются выпрямление лордоза, гипертрофия поперечных отростков С7 с рудиментарными ребрами.
ЭМГ мышц рук и ног: изменения типичны для мышечного уровня поражения.
Биопсия левой четырехглавой мышцы бедра: в мышечном биоптате обнаружено уменьшение количества волокон в мышечных пучках, диффузная разнокалиберность мышечных волокон с округлением контуров, дистрофическими и атрофическими изменениями в некоторых из них. Общая активность окислительно-восстановительных ферментов высокая, дифференцировка на типы волокон при обработке на гликоген, сорбитдегидрогеназу нечеткая. Имеется разрастание соединительной ткани.
Заключение: морфологическая картина в мышце характерна для прогрессирующих мышечных дистрофий.
ФДФ-альдолаза сыворотки крови — 49 ЕД, ЛДГ — 575 ЕД, ACT — 209 ЕД. Креатин мочи — 1,52 ммоль/сут, креатинин — 3,08 ммоль/сут.
Кровь: общий белок — 71 г/л, альбумины — 48 г/л, кальций — 2,36 ммоль/л, фосфор — 1,6 ммоль/л, холестерин — 4,6 ммоль/л, глюкоза — 4,99 ммоль/л, азот мочевины — 9,28 ммоль/л, мочевая кислота — 0,25 ммоль/л, общий билирубин — 6,84 мкмоль/л, щелочная фосфатаза — 125 мЕ/мл, натрий — 143 ммоль/л, калий — 4,4 ммоль/л, хлориды — 105 ммоль/л, СО2 — 25 мэкв/л, АТФ — 0.
Осмотр детским гинекологом: длина и масса тела соответствуют возрасту. Видимых соматических пороков, характерных для синдрома Шерешевского — Тернера, нет. Наружные половые органы сформированы по женскому типу. Отмечаются синехии малых половых губ.
Исследование per rectum: высоко в малом тазу определяется маленькое образование величиной 0, Х 0,5 см, по-видимому, матка с длинной шейкой в виде тяжа, придатки не определяются.
Исследование кариотипа: в лимфоцитах периферической крови выявлено 3,5% клеток с моносомией по Х-хромосоме, в культуре фибробластов кожи обнаружено 20% моносомных по Х-хромосоме клеток. При исследовании соскоба со слизистой щеки хроматиноотрицательные клетки составили 90%.
В родословной данной семьи обращает на себя внимание отсутствие детей мужского пола во II и III поколениях по материнской линии.
Родословная семьи С-вы

Миодистрофии Дюшенна. Обозначения те же, что и на рисунке Родословная семьи Д-вых.
Таким образом, у девочки 5 лет выявлена мягко выраженная картина миодистрофии. Высокий уровень ферментов в сыворотке крови, гипертрофия икроножных мышц, раннее развитие ретракции ахилловых сухожилий, генеалогический анализ, свидетельствующий об отсутствии лиц мужского пола по материнской линии, указывают на наличие миодистрофии Дюшенна. Особенности строения тела девочки, данные гинекологического обследования и, наконец, цитогенетическое исследование дали основание поставить диагноз мозаицизма по Х-хромосоме (ХО/ХХ).
Важно подчеркнуть, что в двух исследованных тканях — лимфоцитах крови и фибробластах кожи — отмечалось различие содержания аномальных клеток. Мягкость течения миодистрофии Дюшенна объясняется тем, что в данном случае имеется мозаицизм ХО/ХХ, а не «чистый» синдром Шерешевского — Тернера.
«Нервно-мышечные болезни»,
Б.М.Гехт, Н.А.Ильина
Причины и симптомы атрофии мышц ног, бедра и голени
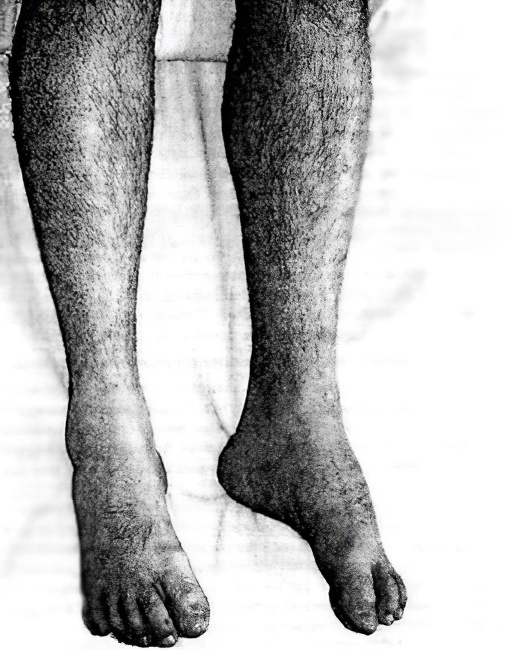
Вследствие патологических изменений в организме человека скелетная мышца начинает истончаться, деформироваться, затем происходит её замещение соединительной тканью, неспособной к сокращению, то есть происходит атрофия мышц. Как результат снижается двигательная способность пораженной мышцы, а при значительном её перерождении происходит полный паралич, больной теряет способность самостоятельно передвигаться.
Причины атрофии мышц ног, бедра и голени
Причин для развития атрофии мышц нижних конечностей может быть несколько:
– снижение метаболизма и старение организма с возрастом;
– как результат заболеваний эндокринной системы и гормонального сбоя в организме;
– хронические заболевания пищеварительного тракта, соединительной ткани;
– нарушение регуляции мышечного тонуса при поражении периферических нервов, полиневритах, как проявление осложнений некоторых инфекционных и паразитарных заболеваний, хронических отравлениях;
– плохая наследственность – врожденная ферментопатия или генетические нарушения;
– неполноценное, недостаточное питание;
– как посттравматические осложнения или при постоянной физической нагрузке.
Заболевания, связанные с атрофией мышц, как правило, относятся к редким врождённым генетическим заболеваниям, которые проявляться начинают уже в детстве.
Симптомы атрофии мышц ног, бедра и голени
В самом начале заболевания характерным симптомом является быстрая утомляемость в ногах, мышечная слабость при длительной физической нагрузке. Заметно увеличиваются икроножные мышцы. Атрофия обычно начинается с проксимальных (ближайших к телу) групп мышц нижних конечностей. Проявляется это в ограничении двигательной функции ног – больному тяжело подниматься по лестнице и вставать из горизонтального положения. Со временем изменяется походка.
Атрофия мышц развивается медленно и длится годами. Болезнь может распространяться как на одну, так и на обе стороны; процесс может быть как симметричным, так и ассиметричным. Все проявления зависят от причин и формы заболевания, возраста и состояния организма пациента. Клинические проявления заключаются в нарастающей слабости в нижних конечностях, появляется дрожание. Больные испытывают неприятные ощущения, чувство ползанья мурашек под кожей.
Самым характерным признаком развивающейся мышечной атрофии является уменьшение в объёме пораженной мышцы, что замечается даже самим больным на ранней стадии заболевания. Все труднее становится передвигаться без посторонней помощи, особенно тяжело подниматься и спускаться по лестнице. Заболевание протекает хронически, отмечаются периоды рецидивов (с сильными болями в пораженной мышце) и ремиссий с незначительным угасанием симптоматики.
Для первичной формы атрофии мышц характерно поражение самой мышцы, её двигательных нейронов, обусловленное неблагоприятной наследственностью или рядом других причин – травмами, ушибами, физическим перенапряжением. Больной очень быстро утомляется, мускулатура теряет тонус, характерны непроизвольные подергивания конечностей.
Вторичное поражение мышечной ткани нижних конечностей называется невральной амиотрофией, наиболее часто является последствием травм или перенесенных инфекционных заболеваний, как следствие генетической патологии. При этом страдают мышцы голеней и стоп, происходит их деформация. Стопа как будто бы висит, и чтобы не цепляться ею за пол человек начинает высоко поднимать колени при ходьбе. По мере прогрессирования и распространения процесса, атрофия мышц с ног переходит на кисти рук и предплечья.
Миотония, сцепленная с полом, протекающая с атрофией мышц ног
Псевдогипертрофическая форма Дюшенна относится к самым распространенным формам миопатии, сцепленной с полом. Заболевание встречается только у мальчиков. Ранние симптомы патологии появляются в первые пять лет жизни ребенка. К характерным симптомам относится атрофия мышц ног и мышц тазового пояса. Рано развиваются псевдогипертрофии, особенно в области икроножных мышц, реже поражаются дельтовидные мышцы. Появляются также концевые атрофии мышц, ретракции сухожилий, главным образом ахиллова, пропадают рефлексы, больше всего это заметно при проверке коленных рефлексов. Ребенок с трудом поднимается вверх по лестнице, опираясь при ходьбе руками о бедра, не может прыгать, ему тяжело подниматься с пола. Постепенно развивается слабость, атрофируются мышцы плечевого пояса, а спустя некоторое время ребенок не может подняться с постели. Среди поздних проявлений заболеваний можно отметить появление контрактуры, причиной которой становится ретракция сухожилий, формирование «конской» стопы.
Как правило, дети с данным врожденным генетическим заболеванием не доживают до 14 лет.
Патология сопровождается также изменениями со стороны сердечной мышцы, поражается головной мозг, ребенок отстает в развитии. Слабость дыхательных мышц становится причиной плохой вентиляции легких, что способствует развитию пневмонии. Течение пневмонии осложняется слабостью сердечной мышцы, что является самой распространенной причиной смерти пациентов. Для формы Дюшенна характерно плейотропное влияние патологического гена.
В середине ХХ века Беккером был описан доброкачественный вариант миопатии, сцепленной с полом, эта форма заболевания носит его имя. Первые симптомы патологии появляются после 20 лет. На начальном этапе заметна псевдогипертрофия икроножных мышц. Атрофия мышц ног развивается медленно, постепенно охватывая мышцы тазового пояса и бедер. Интеллект при этой форме сохраняется. Эти разновидности заболевания характеризуются повреждениями различных генов, располагающихся в двух локусах половой Х-хромосомы, являясь генокопиями. В одной семье сразу две формы заболевания не встречаются.
Диагностика атрофии мышц ног, бедра и голени
Для того, чтобы диагностировать атрофию мышц необходимо собрать тщательный анамнез, в том числе и узнать о наследственных и хронических заболеваниях. Назначается развернутый анализ крови с обязательным определением СОЭ, глюкозы, печеночных проб. Обязательна электромиография и иногда биопсия нервных клеток, а также исследование нервной проводимости. При наличии в анамнезе хронических заболеваний или перенесенных инфекционных, по показаниям проводится дополнительное обследование.
Лечение атрофии мышц ног, бедра и голени
При выборе лечения основное внимание уделяют причинам, из-за которых развилось заболевание. Учитывается возраст пациента, распространенность и тяжесть патологического процесса. Медикаментозное лечение, проводимое курсами, способно приостановить процесс и даже приводит к некоторым улучшениям. Немаловажную роль играет назначение физиотерапевтического лечения, лечебного массажа, электролечения, лечебной гимнастики. Также при лечении атрофии мышц нередко практикуют переливание крови. Соблюдение всех рекомендаций позволяет больным вести практически нормальный образ жизни на протяжении длительного времени.

Эксперт-редактор: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность – “Лечебное дело” в 1991 году, в 1993 году “Профессиональные болезни”, в 1996 году “Терапия”.
Мышечная дистрофия поясноконечностная
OMIM 253600
Наша команда профессионалов ответит на ваши вопросы
В 1954 году доктором Уалтоном и доктором Натрассом был впервые введен термин поясно-конечностые мышечные дистрофии (ПКМД илиLGMD), однако четкого понимания механизма заболевания не было. Это был универсальный термин, который использовали достаточно широко для разграничения людей с преобладающей конечностно-поясной слабостью от людей с другими типами дистрофий, таких, как Дюшена, Ландузи-Дежерина и др.
Поясно-конечностные мышечные дистрофии (ПКМД) – группа прогрессирующих мышечных дистрофий, для которых характерно изолированное или преимущественное поражение мышц плечевого и тазового поясов конечностей. Типичными клиническими проявлениями этой группы заболевания являются нарушения походки (переваливающаяся или «утиная» походка), «осиная» талия, приемы Говерса (подъем лесенкой из положения на корточках), гиперлордоз в поясничном отделе позвоночника, «крыловидные» лопатки, симптом «дряблых надплечий» (при попытке приподнять больного за подмышечные области, его плечи свободно поднимаются вверх, а голова как бы проваливается между ними), сухожильная гипорефлексия, мышечная гипотония и гипотрофия. Мышцы лица, как правило, не поражаются. Слабость проксимальных мышц может начаться либо в верхних конечностях, либо в нижних, но обычно при прогрессировании патологического процесса поражаются все четыре конечности. Мышечная слабость может проявиться уже в детстве (в возрасте до пяти лет) или же позже: например, на 3-м десятилетии жизни; иногда она сопровождается псевдогипертрофией икроножных и других мышц, контрактуры выявляются редко.
После первых проявлений заболевания больной может сохранить способность ходить более 20 лет. Мужчины и женщины болеют в равной мере. Некоторые врачи отмечают, что если болезнь появилась в детстве, то она прогрессирует быстрее и разрушительнее; если начало пришлось на юность или взрослый возраст, то обычно болезнь прогрессирует медленнее.
У некоторых больных в патологический процесс вовлекается сердце, но это бывает не так часто, как при других видах миодистрофий. Проблемы с сердцем могут проявиться в виде кардиомиопатий (слабость сердечной мышцы) или аритмий. В отдельных случаях заболевание само по себе может проявляться как кардиомиопатия.
По истечении тридцати или более лет к основным признакам болезни присоединяется дыхательная недостаточность. Боль не является характерной составляющей болезни. Но ограниченная подвижность иногда приводит к болезненности мышц и суставов. Интеллектуальные функции не страдают. Для большинства нозологических форм поясно-конечностных прогрессирующих мышечных дистрофий характерно повышение активности креатинфосфокиназы в плазме крови больных, которое может выявляться еще на доклинической стадии. Увеличение активности этого фермента может выявляться и у гетерозиготных носителей мутации в том или ином гене, которое, однако, не достигает такой степени, как у больных.
На ЭМГ и в мышечном биоптате выявляются признаки миопатии, мышцы истончены, часть волокон замещена жировой и соединительной тканью. В саркоплазме выявляются очаги фокального некроза. Ядра мышечных волокон центрально смещены, располагаются рядами или цепочками, вакуолизированы, с выраженным ядрышком. На поздних стадиях волокна теряют поперечную исчерченность, фрагментированы; иногда обнаруживаются только остатки миофибрилл.
При дифференциальной диагностике необходимо исключить воспалительные и метаболические миопатии, а также фенотипически сходные спинальные мышечные атрофии.
Частота всех поясно-конечностных мышечных дистрофий колеблется в различных популяциях от 5 до 70 больных на 1 миллион населения.В зависимости от типа наследования поясно-конечностные мышечные дистрофии разделяют на два типа.
К 1 типу относят нозологические формы с аутосомно-доминантным типом наследования. Для всех заболеваний этой группы известна локализация генов на хромосомах и для трех из них идентифицированы белковые продукты.
Ко второму типу относят формы заболевания, наследующиеся аутосомно- рецессивно. Первое описание ПКМД с аутосомно-рецессивным типом наследования проведено Kloepfer и Tally в 1958 году. Для большинства заболеваний этой группы идентифицированы гены, описаны основные типы их патологических мутаций и известен белковый продукт и его основные функции. На данный момент выделено 16 нозологических форм ПКМД с аутосомно-рецессивным наследованием и 7 с аутосомно-доминантным (Таблица 1), и их поиск продолжается.
Аутосомно-рецессивные формы ПКМД являются более распространенными, чем аутосомно- доминантные, которые составляют около 10% всех ПКМД. Различные группы населения часто имеют разные частоты различных типов ПКМД. Среди аутосомно-рецессивных форм наиболее распространены ПКМД 2A (от 30 % в Бразилии до 80% среди басков Испании от всех выявленных случаев) и ПКМД 2B (от 20% до 40% всех случаев). На группу саркогликанопатий (ПКМД 2C-2F) приходится еще около 20-25% всех случаев ПКМД, эта группа характеризуется тяжелым течением заболевания. Как и в случае с другими ПКМД, различные саркогликанопатии с различной частотой встречаются в разных популяциях. ПКМД 2C широко распространена в Тунисе; ПКМД 2D распространена в Европе, Соединенных Штатах и Бразилии, а ПКМД 2E и ПКМД 2F широко распространены в Бразилии. ПКМД 2I довольно распространена, особенно среди жителей Северной Европы. Недавние исследования показали, что больные с мутациями в этом гене составляют 6-38% случаев ПКМД. Остальные аутосомно-рецессивные формы ПКМД встречаются редко, и часто наблюдаются в изолированных группах населения.
В ООО «Центр Молекулярной Генетики» методом прямого автоматического секвенирования всей кодирующей последовательности проводится диагностика следующих форм ПКМД (LGMD1A, LGMD1B, LGMD1С, LGMD2А, LGMD2С, LGMD2D, LGMD2E, LGMD2F, LGMD2К, LGMD2I, LGMD2М, LGMD2L). Данные формы выделены в таблице синим цветом.
Для LGMD 2I типа описаны частые мутации с.826C>A, с.229С>T, которые встречаются хотя бы на одной из хромосом более чем у 80% больных из РФ.
Для LGMD 2A типа описаны частые мутации с.550delA, с.598_612del15, которые встречаются хотя бы на одной из хромосом более чем у 83% больных из РФ.
По литературным данным для LGMD 2D типа описаны частые мутации, характерные для больных из РФ: c.229C>T и c.271G>A.
По литературным данным для LGMD 2L типа описаны частые мутации, характерные для европейцев: c.191dupA и c.2272C>T.
В ООО «Центр Молекулярной Генетики» проводится анализ частых мутаций с.826С>A, с.229С>T и замены с.341С>T гена FKRP, c.191dupA и c.2272C>T гена ANO5 и c.229C>T и c.271G>A гена SGCA , с.550delA и с.598_612del15 гена CAPN3.
Таблица 1. Гены, ответственные за развитие различных форм ПКМД
Тип
заболе-
вания
OMIM
Тип
наследо-
вания